Background
Inherited metabolic disorders (IMDs) are a heterogeneous group of disorders, each caused by deficient activity of a single enzyme in a pathway of intermediary metabolism. Interruption of different pathways can lead to accumulation of pathological/toxic products or a deficiency of products essential for health, causing systemic multi organ damage and in most cases long-term physical or learning disability. It is estimated there are between 6,000 and 8,000 individual rare diseases. Although each individual disease is rare, the sheer number of individual rare diseases results in one in 17 of the population being affected.1
Newborn screening for IMDs uses an elute and shoot process that is rapid, relatively cheap and high throughput. This versatile method is advantageous as many analytes can be detected in a short time from very small quantities of sample. However, it has limitations as some metabolites are not always screened for a specific disease, and the technique cannot measure different compounds of the same molecular weight.
In 2019, genetic or heritable conditions are thought to have been responsible for:2
- 30% of deaths <1 year old
- 18% of deaths for ages 1–5 years old
- 11% of deaths for ages 5–15 years old
- 30% of all childhood admissions to hospital
- Two-thirds of all disability.
The UK newborn blood spot (NBS) screening programme started in the 1950s when phenylketonuria (PKU) was screened for using the Phenestix test, which was replaced with the dried bloodspot (DBS) system developed by Robert Guthrie in the late 1960s. The use of bloodspots on Guthrie cards is still in use to this day. In 1969, screening was centralised by the Department for Health and regional hub screening laboratories were established. Screening was devolved across regions, with marked variation in the scope of disorders until 1996 when the UK National Screening Committee (NSC) was launched. The NSC chose 20 criteria, based on adapted Wilson & Junger principles, against which screening for disease can be assessed.3
With the advent of tandem mass spectrometry (MS/MS or TMS) technology, screening for IMDs became faster, more sensitive and expandable. Mass spectrometry is a technique used for both identifying and quantifying compounds. Samples are ionised, the charged molecules are extracted into the analyser, separated by their mass-to-charge ratios and quantified by their relative intensity as they emerge. Newborn screening for IMDs uses an elute and shoot process that is rapid, relatively cheap and high throughput. This versatile method is advantageous as many analytes can be detected in a short time from very small quantities of sample. However, it has limitations as some metabolites are not always screened for a specific disease, and the technique cannot measure different compounds of the same molecular weight.
Currently, the UK NBS programme screens for six IMDs alongside three other disorders (congenital hypothyroidism, sickle disease and cystic fibrosis). A capital investment, in terms of tandem mass spectrometers, for medium chain acyl-CoA dehydrogenase deficiency (MCADD) screening took place in 2007. In 2008, screening was rolled out nationally after a two-year pilot study met the rigorous criteria set out by the NSC. PKU was also moved over to tandem MS/MS. This was followed by an ambitious pilot in 2012, covering a further five disorders, which could all easily be screened by existing MS/MS technology.
Of these five disorders, four were then included in the NBS bloodspot programme: isovaleric acidaemia (IVA), gluatric aciduria type 1 (GA1), homocystinuria (HCU) and maple syrup urine disease (MSUD). The addition of multiple other analytes represents a small incremental increase in cost, and screening for the four additional conditions is cost saving. The incremental net benefit for all four conditions, at a threshold of £25,000 per quality-adjusted life year, was between £0.46 for IVA and £5.94 for GA1.4
| Table 1: Comparison of screening in the UK versus the Netherlands. | ||
|---|---|---|
| Disorder | UK | Netherlands |
| Cystic fibrosis | Cystic fibrosis | |
| Haemoglobinopathies | Sickle cell disease Beta thalassemia major |
Sickle cell disease Haemoglobin H disease (a form of alpha thalassemia) Beta thalassemia major |
| Endocrinology | Congenital hypothyroidism | Congenital hypothyroidism Congenital adrenal hyperplasia |
| Amino acid disorders | PKU MSUD HCU |
PKU MSUD HCU Tyrosinemia type 1 |
| Other metabolic disorders | IVA GA1 |
IVA GA1 3-Methylcrotonyl-CoA carboxylase deficiency Biotinidase deficiency Galactosemia HMG-CoA lyase deficiency 1 |
| Fatty acid oxidation defect | MCADD | MCADD Multiple acyl-CoA dehydrogenase deficiency Very long-chain acyl-CoA dehydrogenase deficiency Carnitine transporter (OCTN2) deficiency 2 |
|
|
||
Comparing IMD screening in the UK to other countries
In the UK, all babies under a year of age are eligible for NBS screening for all nine conditions. Blood is collected via a heel prick test on day five of life and blotted onto a Guthrie card, which is sent to one of 13 laboratories with results released within a maximum of 72 hours upon receipt of the sample. Babies who screen positive are immediately referred to local clinical metabolic medicine teams and seen the same day for complete clinical assessment and confirmatory diagnostic testing alongside appropriate treatment. Families who have an affected child and go on to have further children are offered pre-implantation genetic diagnosis or, where they choose not to receive this specialist IVF treatment, the siblings of known cases are screened rapidly on day one of life for the known metabolic condition.
Across Europe there are significant variations in screening protocols, with countries screening between one to 30 different conditions with unique inclusion criteria.
The USA has taken a very different approach to choosing conditions for newborn screening. Inherited disorders were individually assessed against selection criteria, but also treated as a whole group. Some untreated conditions were included on grounds that screening ‘might provide benefit to society’ through reducing cost of diagnosis per condition and generating research benefit while maximising use of current technology.
29 core conditions were recommended by the American College of Medical Genetics (all of which have been adopted), and 25 optional secondary conditions that individual states can decide to include in their panels.5 Owing to the complex healthcare system in the USA, although patients may be given a screening diagnosis, those without insurance may struggle to access appropriate healthcare for their condition and may be refused insurance throughout their life. Many of the conditions screened for in the US have no specific treatment yet. This approach may be of less benefit to individual families, who may then have to endure the knowledge of their child’s often limited lifespan while being unable to benefit from further specific treatment.
Across Europe there are significant variations in screening protocols, with countries screening between one to 30 different conditions with unique inclusion criteria. An example of a screening protocol in the Netherlands compared with the UK can be seen in Table 1.
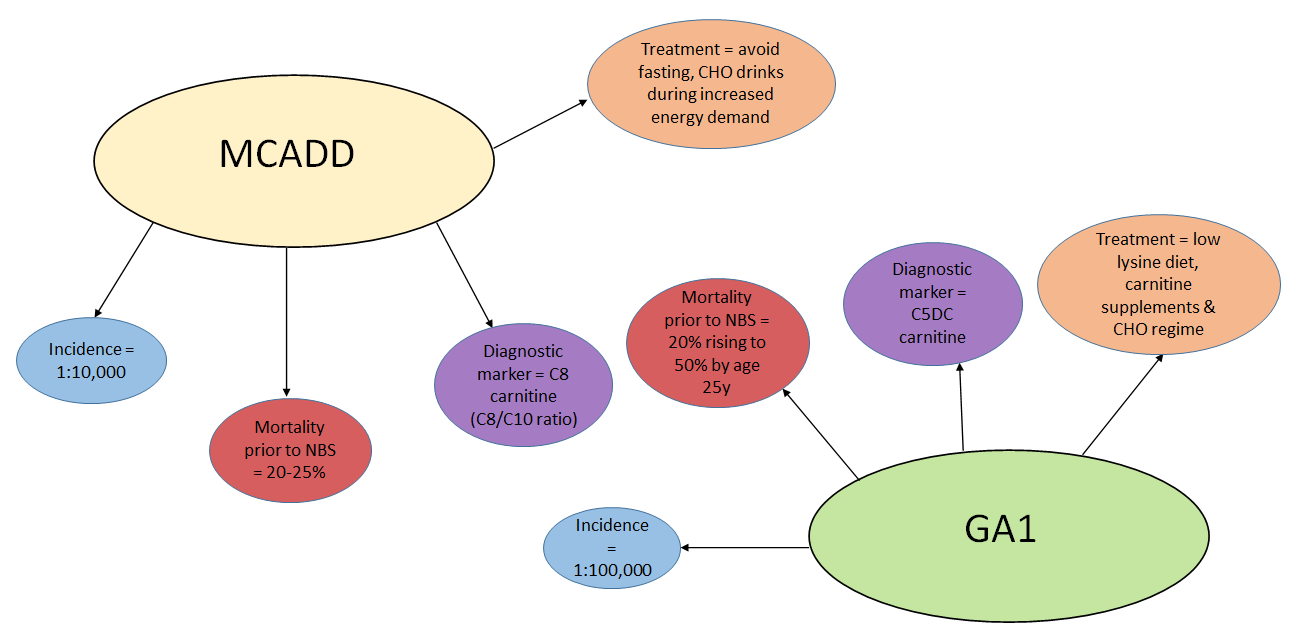
In the UK, the screening program has been well received, with only 2.8 in 1000 parents refusing screening.6 The concerns of false positives causing undue stress and harm to families remains a challenge, particularly for IVA where maternal use of pivalate antibiotics causes artefactually elevated C5 carnitine. However, children correctly identified by screening benefit significantly through reduction in morbidity and mortality (see Figure 1).
Future directions for NBS
In 2019, the then Health Secretary, Matt Hancock, and Genomics England announced a pilot to allow parents to opt into having their newborn babies’ whole genome sequence tested. This assesses the risk of developing health conditions over their lifespan. The assumption was that many lives would be saved knowing the risk of disease. However, large-scale studies in the USA have shown that the number of false negatives and false positives are greater than by the current method of MS/MS. Furthermore, as there is still uncertainty about the function of many genes and whether some variants identified in them are pathological, functional testing through metabolome profiling is still more successful. An embedded hybrid approach is more likely to be beneficial in the long term.
Currently, the NBS programme is undertaking a pilot to assess the addition of a tenth disease – although not an additional metabolic condition – to add to the nine conditions already screened for. Using the DBS system already in place, laboratories will screen for severe combined immunodeficiency,a genetic disorder affecting the development of functional T cells and B cells in infants which, if left untreated, results in repeated severe infections and death within the first few years of life. Currently,the standard treatment is haematopoetic stem cell transplant, with outcomes that seem to be associated with a younger age of transplant and being free from infections.
There are other metabolic disorders likely to be added to the UK NBS programme over the next 10–15 years. Tyrosinemia type 1 is an autosomal recessive disorder due to mutations in the gene encoding fumarylacetoacetase. Screening for succinylacetone (one of the main metabolites) on DBS by MS/MS has been implemented in many countries and could easily be admitted to the existing screening programme. Tyrosinemia type 1 can present early, with liver dysfunction, growth failure and rickets, and/or later with similar symptoms as well as with neurological crises. Untreated, they can both lead to death by ten years of age. Treatment with nitisinone is effective and can prevent most of the long-term effects, justifying its inclusion in the panel.
Biotinidase deficiency is another metabolic disorder that could be added to the existing programme using MS/MS technology. Deficiency of the enzyme biotinidase leads to a loss of recycling of the vitamin biotin, which is used as a cofactor in many pathways within the body. Symptoms including seizures, hearing loss, breathing difficulties, hair loss and ataxia can present early in life. Treatment is simple and effective.
Finally, lysosomal storage disorders (LSDs) may be included. These disorders are characterised by enzymatic deficiencies that cause build-up of substrates and by-products within lysosomes, leading to multi organ dysfunction and often early death if left untreated. For LSDs to be added to the UK NBS programme, a methodology will need to be optimised to allow use of DBS and increased throughput within a reasonable timeframe. Traditional diagnosis of LSDs involves lengthy enzyme assays to assess activity. However, recently, digital microfluidic fluorimetry has been developed to multiplex enzymatic assays with a same day turn-around on a simple to use platform. Furthermore, there are already diagnostic tests available to confirm screening positives and many disorders now have enzyme replacement therapies for treatment, which, allied with early diagnosis, result in significant quality-of-life and lifespan improvements.
The UK NBS programme has often received criticism for only screening a small number of conditions, especially compared with other countries. However, the UK has developed an exceptionally well regulated and universally available system, which screens upwards of 750,000 of infants each year with rapidly available results.6
Further information is available at: http://newborn-screening.org/site/index.asp.
Author(s)

Raphael Buttigieg



