The application of nucleic acid sequencing and sequence-dependent detection methods have been used for the diagnosis and management of viral infections for decades. Viruses, whose nucleic acid is a minor component of the total nucleic acid in a clinical sample and which often cannot be easily rescued in cell culture, also have genomes that vary in nature consisting of single- or double-stranded (ss or ds) ribonucleic acid (RNA) or deoxyribonucleic acid (DNA). Thus enrichment of viral genetic material is required prior to DNA sequencing. Historically, this has been accomplished by target-specific amplification using polymerase chain reaction (PCR), preceded by reverse transcription (RT) for RNA viruses, and followed by DNA sequencing using Sanger-based technology. However, in the past decade great advances in sequencing technologies are now predicted to transform how clinical microbiology laboratories diagnose and manage infectious diseases.
The advent of next-generation sequencing (NGS) that enables massively parallel sequencing of millions of DNA templates in a single reaction means that whole genome sequencing (WGS) of hundreds of microbial samples can be accomplished in a matter of hours and in theory at very low cost. The techniques and data generated have a wide range of applications in the management of viral infections, from identifying and characterising sources of infection to determining the effect of host immune response, treatment and intervention strategies.
Here, we explore these new technologies and briefly discuss some of the added value of using them for the clinical diagnosis and management of viral infections, with particular emphasis on the blood-borne viruses human immunodeficiency virus (HIV) and hepatitis C virus (HCV).
Viral NGS methods
Sequencing platforms normally require ample amounts (nanograms or more) of dsDNA as starting material, therefore most viral genomes have to be amplified and converted to dsDNA prior to sequencing. Sanger-based methods achieve this through target-specific amplification followed by sequencing of the resulting amplicons using two or more overlapping primers in multiple reactions that cover each nucleotide at least once in both the forward and reverse directions.
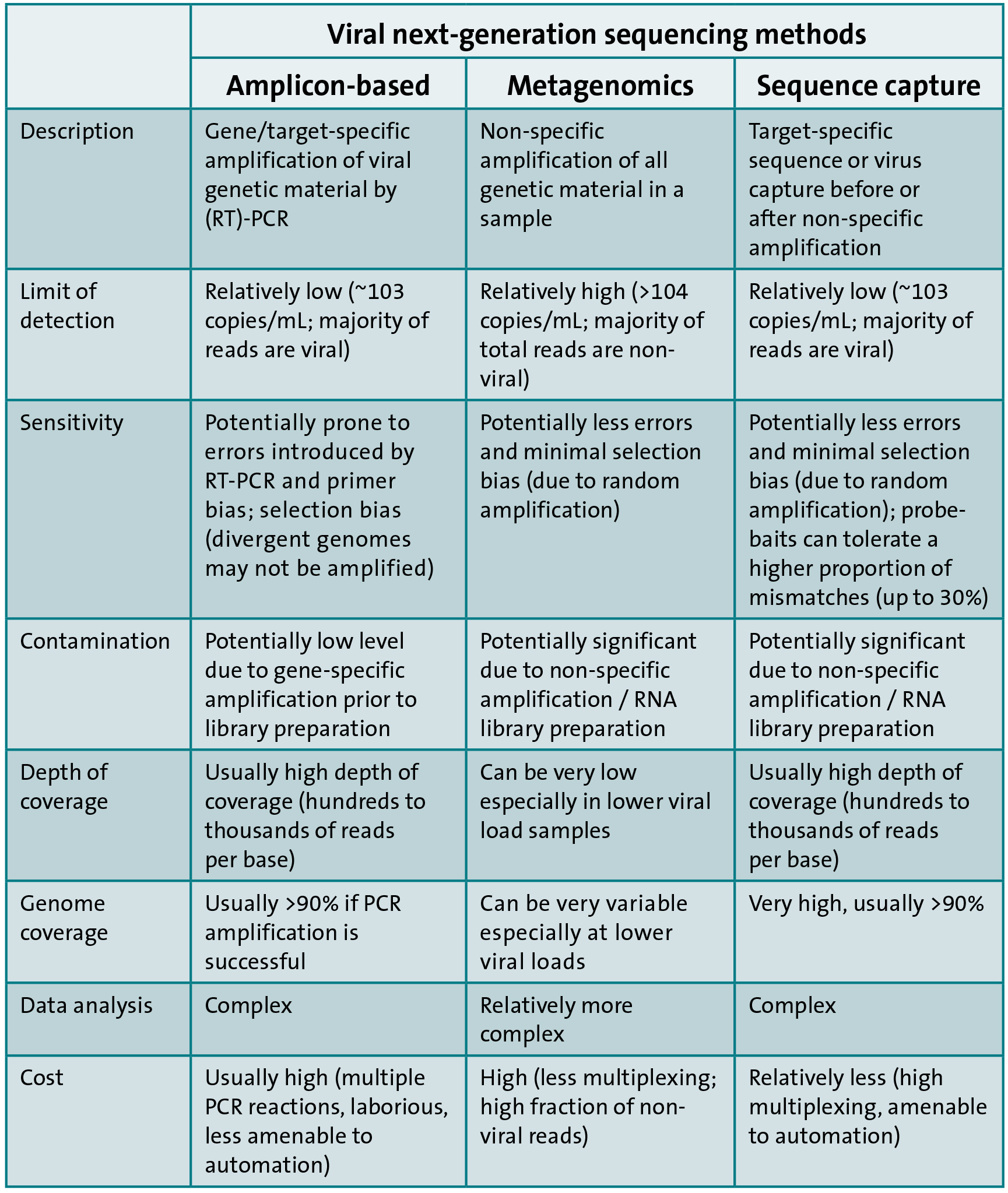
In contrast, three main approaches exist for sequencing viral genetic material by NGS. The first approach is similar to Sanger-based sequencing and involves target-specific amplification followed by library preparation and NGS. The second approach is metagenomic, which involves non-specific amplification of all genetic material (pathogen and host) within a sample followed by NGS. The last approach involves enrichment of viral genetic material by sequence or virion capture prior or subsequent to DNA amplification and library preparation followed by NGS. The advantages and disadvantages of each approach are highlighted in Table 1.
A comprehensive collaborative study to compare the three approaches for the sequencing of full-length HCV genomes showed that sequence capture approaches are the most cost-effective and amenable to fully automated high-throughput workflows. In addition, the probe baits used for sequence capture can be designed to target multiple viruses and this can allow a unified syndromic capture system, e.g. for blood-borne viruses. This is akin to multiplex real-time PCR assays but with the added value of genomic data to enable the characterisation of the infecting viruses and for other public health purposes. On the other hand, metagenomic-based approaches have the potential to detect unknown causes of infection a priori.
Specific applications of genomics in clinical virology
Detection and monitoring of antiviral resistance
Detection of drug resistance in plasma-borne viruses using DNA sequencing has been a mainstay for the clinical management of HIV-infected patients for decades. This method is reliable and fast compared to the alternative, gold standard, cell-based phenotyping in culture and is also used to manage other viral infections that are treated with antivirals including HCV, hepatitis B virus, influenza and herpesviruses. A prerequisite of genotypic antiviral resistance testing is the availability of a robust sequence database for the prediction of drug susceptibility from genomic data. Several exist for HIV, such as the Stanford HIV drug resistance database (hivdb.stanford.edu/), which is publicly accessible and regularly updated with new mutations and drugs. Sanger-based methods continue to be routinely used for this purpose and for HIV this involves the amplification of three separate genomic regions: protease-reverse transcriptase, integrase and V3 loop of envelope. Thus one advantage of WGS is that it can undertake these tests in a single reaction. This would inform the most appropriate and cost-effective regimen on which to start a patient soon after diagnosis. Additionally, the high sensitivity of NGS offers the ability to detect low frequency drug-resistant variants in mixed virus populations at frequencies down to 1% or lower compared to Sanger-based methods that have a limit of detection >20% variant frequency. The low frequency variants can rapidly become the majority population culminating in virologic failure upon initiation of antiviral therapy. However, the clinical significance of the low frequency drug-resistant variants is not yet fully understood.
Characterisation of viral genotypes
Defining the genotype of an infecting virus is important for the management of some viral infections, exemplified by HCV, a virus which is highly diverse and divided into seven genotypes that are further subdivided into over 65 subtypes. The effectiveness of HCV treatment, based on either interferon or the new direct-acting antivirals (DAAs), remains genotype-dependent. For example, interferon-based regimens are less successful in genotype 1 than genotype 2 and 3 infections, whereas first-generation protease inhibitors are most effective against genotype 1. Thus the determination of infecting genotype is an integral part of the clinical management of HCV-infected patients and a useful indicator of prognosis and duration of therapy. Several methods have been developed for HCV genotyping that involve amplification of a region of the HCV genome to distinguish the genotype or subtype by restriction fragment length polymorphism (RFLP), probe hybridisation, real-time PCR or sequence analysis. The target of the assays is usually a conserved region of the HCV genome such as 5’-UTR, core or NS5b, that permit pan-genotypic primer design but conversely afford significant variation in the inter-primer region to differentiate genotypes and subtypes. Sequence analysis assays have been shown to be the most accurate, whilst the commonly used methods employing single base polymorphism (SNP) can assign the wrong subtype or genotype that adversely affects treatment outcome. WGS, by its nature, can refine the assignment of genotype and detect mixed genotype infections or rare recombinant forms. Thus, for HCV, the use of WGS would be cost-effective and significantly enhance the clinical diagnostic workflow by combining the multiple current assays used for genotyping and drug resistance detection into a single assay whilst enhancing the granularity of genotype assignment.
Emerging uses of genomics in clinical virology
Personalised medicine
Also called stratified or precision medicine, this is becoming a useful approach for the treatment and prevention of diseases. For infectious diseases, this involves targeting specific treatments and preventions at individuals more likely to benefit by using pathogen and host genomic data in combination with other clinical data. The approach is already part of the management of some viral infections and is bound to become commonplace with the increase in the availability of genomic data and a better understanding of host-pathogen-therapy interactions. For example, a C/T genetic polymorphism in the host interleukin-28B gene is a strong predictor of viral clearance in HCV-infected individuals and is often used to inform interferon-based HCV therapy management. Host genomic data can also be used to maximise the effects of a particular therapy by reducing the occurrence of adverse incidents. An example is the screening for the HLA-B*5701 allele, which has been shown to be linked with an increased risk to a hypersensitivity reaction against the antiretroviral drug, Abacavir. The increase in genomic and linked metadata will allow the development of predictive treatment stratification models that will improve clinical decision making and is a focus of several studies such as the MRC-funded ‘Stratified Medicine to Optimise Treatment for HCV’ (STOP-HCV, www.stop-hcv.ox.ac.uk). The use of genomics to stratify treatment or interventions raises ethical issues that require careful consideration to safeguard patients’ autonomy and privacy whilst ensuring maximum impact and benefit to the general public. For example, the withholding of treatment from patients with a particular genotype can be considered unethical as it could be argued that all affected patients should have an equal chance at receiving the same treatment, especially as genotype-disease associations are often not absolute.
Near real-time genomics for transmission investigation and infection control
Viral genomics has been used for decades to determine the sources of infections as part of a public health strategy for infection control. The new sequencing technologies allow the rapid generation of WGS data which, when linked to clinical, epidemiological, behavioural and social network data, can provide refined and near real-time evidence to aid the early detection of outbreaks and improved infection control. For example, the approach is being touted as a way to control the increasing levels of HIV-1 transmissions, which is thought to mainly be due to undiagnosed infections. It has been proposed that sequencing of newly diagnosed individuals, followed by adding their sequence to an active phylogenetic tree containing sequences from others in a particular geographic region generated as part of routine genotyping, would help determine if the sequence is part of a transmission network. The expansion of a transmission cluster would then trigger a public health response. Conversely, if a newly diagnosed sequence falls outside a cluster, this could trigger enhanced contact tracing to potentially identify undiagnosed individuals and help bring them into clinical care, thereby reducing onward transmission. In addition, the distribution of shared minority variants between individuals in a potential transmission cluster, that can be determined using NGS technologies, can shed light on the dynamics of transmission events and further refine transmission investigations. However, such approaches seeking to identify sources and dynamics of transmission events are controversial and raise considerable ethical and legal questions, which need careful consideration to avoid the misuse or misinterpretation of the information gathered.
Vaccine design and development
Immunisation is one of the most cost-effective preventive strategies against infectious diseases. Due to the diversity of viruses, the lack of cell culture and animal models for some viruses, genomics provides a promising approach for the design and development of vaccines. For example, owing to the rapid changes exhibited by the influenza virus, the vaccine has to be reformulated on a yearly basis. A WHO global influenza surveillance network decides on the strains to include in each year’s vaccine – a decision informed by the surveillance of circulating isolates worldwide followed by antigenic characterisation. Incorporating genomic data in this process has been shown to enhance significantly the understanding of the varying antigenic patterns and the evolution of the virus, thereby improving vaccine effectiveness.
Challenges
A significant challenge of using WGS diagnostic tests in the clinical pathway is how to deliver technical and clinical validation, as well as setting the standards for quality assurance and governance. The process has to cover not only the ‘wet’ laboratory process but also the bioinformatics pipeline, reporting and data storage, transfer and access. Clinical microbiology laboratories are learning from pioneering work done by clinical human genetics laboratories that have established standards for use of NGS tests in cancer and inherited diseases. However, owing to the unique challenges encountered in undertaking WGS for microbiology, especially for virology, specific and distinct standards have to be formulated. The setting of technical and clinical cut-offs for calling of minor variants need to be empirically and statistically determined, using well-defined reference material and well-characterised clinical cohorts. The high diversity exhibited by viruses makes the assembly of viral WG from short-read NGS data and the detection of contaminants problematic and the implementation of a fully automated, version-controlled, end-to-end viral WGS workflow challenging. It might be that targeted NGS aimed at relevant regions, e.g. HIV-1 pol or V3 loop of envelope associated with drug resistance could be employed as a tried and tested approach. Thus, an in-depth evaluation of the effectiveness of viral WGS tests has to be undertaken before introducing them in the clinical pathway to ensure they offer additional advantages on current methods, are fit for purpose and provide added value to patient management.
To date, virology WGS/NGS is rarely employed in the clinical pathway, but is mostly being used as a research, surveillance or reference tool in some PHE and/or NHS laboratories. The exception is the use of targeted NGS as a replacement for Sanger sequencing, e.g. for viral drug-resistance testing. The results are then reported back to attending clinicians at ~20% variant frequency cut-off, the equivalent threshold used for Sanger sequencing and for which the clinical significance is well established. The analysis of minority variants is currently undertaken for research and surveillance purposes. On the other hand, metagenomic approaches have been used in emergency responses, e.g. to characterise the Ebola virus strain and help inform the management of returning infected UK healthcare workers in the 2014 outbreak by PHE laboratories.
Conclusions
There is no doubt that WGS/NGS is going to change how clinical microbiology laboratories function. It is also quite clear that there are still several challenges that need to be addressed before these technologies become commonplace in the virology clinical pathway. However, it is just a matter of time before they replace the long-established Sanger-based methods. Further advances in sequencing technology and bioinformatics – such as the use of long-read technology, RNA sequencing and bench-top or portable sequencers– will further facilitate their assimilation into modern clinical virology laboratories and provide rapid testing close to the patient.
Dr Jean L Mbisa and Professor Richard S Tedder
Virus Reference Department
National Infection Services
Public Health England
Author(s)
